
改性殼聚糖對(duì)硫酸廢水中氯離子的吸附性能
國(guó)內(nèi)外煉鋅的主要方式是濕法煉鋅,采用焙燒-浸出-電積工藝生產(chǎn)鋅。由于煉鋅原料鋅精礦或者鋅二次資源都含有大量的S、Cl等元素,使得在焙燒工藝中產(chǎn)生含氯的二氧化硫煙氣,在“淋洗凈化”煙氣制酸過(guò)程中,氯化物特別容易被水和酸吸收,因此產(chǎn)生了大量含氯的高酸廢水。若未加處理直接回用,水中Cl-會(huì)對(duì)生產(chǎn)設(shè)備造成嚴(yán)重腐蝕。此外,高濃度含氯廢水若排入江河,會(huì)使水質(zhì)惡化,甚至污染飲用水源,對(duì)人體健康造成威脅。近年來(lái),由于礦石中氯含量不斷提高,使得硫酸廢水中的氯含量也越來(lái)越高,含氯硫酸廢水的處理迫在眉睫。
目前,國(guó)內(nèi)外所用去除工業(yè)廢水氯離子的方法主要有沉淀法、離子交換法、膜分離技術(shù)、電吸附、電解法、電滲析和反滲透,但是這些方法對(duì)于高濃度含氯廢水的處理,普遍存在去除效率低、成本高、能耗高、操作方式復(fù)雜、出水難以達(dá)到環(huán)保要求等缺點(diǎn)。現(xiàn)行常用的石灰鋁鹽沉淀法具有成本低、去除效率高優(yōu)點(diǎn),但其主要缺點(diǎn)是產(chǎn)生大量化學(xué)污泥成為固體廢棄物,在固體廢棄物監(jiān)管日益嚴(yán)格的情況下,該方法的使用受到了限制。因此尋找一種價(jià)格低廉、無(wú)二次污染、高效去除酸性冶煉廢水中Cl-的方法具有重要意義。筆者研制了一種Hg2+改性殼聚糖(HgCTS)高效氯離子吸附劑,研究了其對(duì)Cl-的吸附機(jī)理,并探討了吸附劑投加量、溶液初始pH、吸附時(shí)間對(duì)吸附劑除Cl-性能的影響。
1、實(shí)驗(yàn)部分
1.1 試劑與儀器
實(shí)際硫酸廢水選取廣西某冶煉廠硫酸廢水(Cl1050mg/L、SO42-18400mg/L、F255mg/L、Fe3+1140mg/L、Zn2+82400mg/L、pH=3.3)。殼聚糖(CTR)(脫乙酰度≥90.0%)、硫酸汞、濃硫酸、檸檬酸、丁二酸、氯化鉀、硝酸鉀、氫氧化鈉、冰乙酸、戊二醛(體積分?jǐn)?shù)為2.5%)。總離子強(qiáng)度調(diào)節(jié)劑(自配):稱(chēng)取88g硝酸鉀和量取100mL冰乙酸溶于燒杯中,用10g/L氫氧化鈉調(diào)節(jié)pH至5~5.5,定容于1000mL容量瓶中。
SHZ-82A氣浴恒溫振蕩器, DF-101S集熱式恒溫加熱磁力攪拌器, PXSJ-216離子計(jì)、217-01參比電極、PCl-1-01氯離子電極, FA-1604電子天平, FE20-FivepH計(jì), DZF-6020真空干燥箱, Nexus傅立葉變換紅外光譜儀, D8AdvanceX射線(xiàn)衍射儀, Kratos Axis Ultra DLD多功能電子能譜儀;JSM5610LV型掃描電子顯微鏡。
1.2 吸附劑的制備
取檸檬酸0.25g、丁二酸0.15g、硫酸汞0.4g于50mL去離子水中,再加入0.1mL濃硫酸將硫酸汞溶解制得汞配位溶液A;稱(chēng)取2g殼聚糖溶于100mL體積分?jǐn)?shù)為2%的冰乙酸溶液,攪拌5h,直至殼聚糖溶液完全無(wú)氣泡,得到殼聚糖乙酸溶液B。將以上A和B兩份溶液混合后攪拌2h,將得到的凝膠用去離子水反復(fù)洗滌,抽濾后,再置于體積分?jǐn)?shù)為2.5%戊二醛中交聯(lián)30min,洗滌、過(guò)濾,60℃下烘干研磨,制得不溶于水改性的殼聚糖(Hg-CTS)吸附劑。
1.3 實(shí)驗(yàn)方法
吸附實(shí)驗(yàn):在150mL的錐形瓶中加入吸附劑和Cl溶液,在一定的Hg-CTS投加量、溶液初始pH、Cl-初始濃度和吸附時(shí)間等實(shí)驗(yàn)條件下,以150r/min在氣浴恒溫震蕩搖床中震蕩一定時(shí)間后,取上清液用離子計(jì)測(cè)定電位值并通過(guò)標(biāo)準(zhǔn)曲線(xiàn)計(jì)算出殘余Cl濃度。計(jì)算Cl的去除率和吸附量。
1.4 分析方法
Cl-的測(cè)定采用直接電位法,量取反應(yīng)后的上清液5mL于50mL容量瓶中,加入20mL緩沖溶液后定容,用離子計(jì)測(cè)定其電位值,然后由氯離子濃度的對(duì)數(shù)與電位值的校準(zhǔn)曲線(xiàn)計(jì)算出廢水中Cl含量。
2、結(jié)果與討論
2.1 物化特性及微觀結(jié)構(gòu)表征
2.1.1 FI-TR分析CTS、Hg-CTS和Hg-CTS吸附Cl-的FI-TR見(jiàn)圖1。
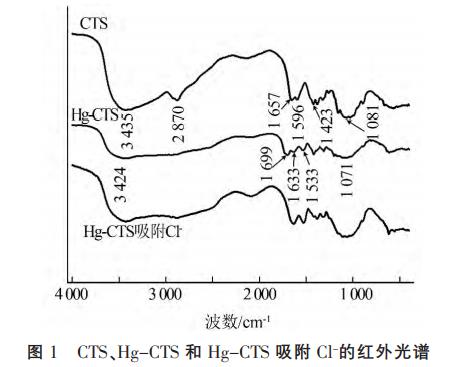
圖1可知,CTS主要吸收帶有:3435cm-1(—OH和—NH2的伸縮振動(dòng))、2870cm-1(—CH和—CH2的伸縮振動(dòng))、1657cm-1(—NH2的彎曲振動(dòng))、1081cm-1(C—O的伸縮振動(dòng))。Hg-CTS中,殼聚糖表面的吸收帶基團(tuán)特征峰發(fā)生了顯著變化,特征峰3435、1657(Ⅰ型酰胺)、1596(Ⅱ型酰胺)、1423(Ⅲ型酰胺)cm-1分別移至3424、1699、1633、1533cm-1,且CTS位于2870cm-1處的—C—H伸縮振動(dòng)吸收峰有所減弱,這可能是由于殼聚糖的游離氨基(—NH2)或游離氨基和二級(jí)羥基(C3—OH)與Hg(Ⅱ)的螯合作用引起的,這和X.Wang等研究的改性殼聚糖吸附Hg(Ⅱ)后峰值變化相符合。對(duì)比CTS,Hg-CTS吸附劑制備成功,對(duì)比Hg-CTS和Hg-CTS吸附Cl后的光譜圖,發(fā)現(xiàn)吸附劑吸附Cl前后的吸收帶基團(tuán)特征峰未發(fā)生明顯變化,說(shuō)明了吸附劑結(jié)構(gòu)比較穩(wěn)定。
2.1.2 XRD分析
殼聚糖XRD譜圖顯示其有兩種不同的晶體形態(tài),均屬于單斜晶系,在2θ分別為10.55°、19.94°處。在汞配位化合物改性殼聚糖后,殼聚糖在10.55°、19.94°處的衍射峰消失了,顯示Hg-CTS是無(wú)定型相,這是由于其處于單層分散狀態(tài)。許多鹽類(lèi)或氧化物具有分散到載體表面形成單層和亞單層的傾向,即當(dāng)負(fù)載物的量低于某一閾值時(shí),呈單層分散狀態(tài),高于此閾值時(shí)出現(xiàn)晶相,X射線(xiàn)檢測(cè)不出活性組分可認(rèn)為處于單層分散狀態(tài)。
2.1.3 XPS分析
為進(jìn)一步研究Hg-CTS吸附劑除氯機(jī)理,CTS和Hg-CTS吸附氯前后的吸附劑被用于XPS分析。掃描能譜全譜圖顯示CTS包含氧、氮、碳元素(O1s、N1s、C1s),而吸附劑Hg-CTS能譜全譜圖除氧、氮、碳元素外,出現(xiàn)了S2p和Hg4f峰,證明殼聚糖被成功改性為載汞吸附劑了。Hg-CTS吸附Cl后能譜全譜圖出現(xiàn)Cl2p峰,證明Cl被吸附于吸附劑表面。
為探討CTS和Hg-CTS吸附Cl前后的吸附劑各個(gè)元素的變化,進(jìn)行了XPS的O1s、N1s、C1s、S2p、Cl2p和Hg4f能譜分析。結(jié)果表明,CTS的O1s光譜的結(jié)合能532.58eV為其表面的—OH、—C—O或結(jié)合水,改性前后基本沒(méi)有發(fā)生變化,但吸附Cl后其強(qiáng)度變小。CTS的N1s光譜的結(jié)合能399.4eV為其表面上NH2基團(tuán)中的N,但Hg-CTS在高結(jié)合能出現(xiàn)一個(gè)明顯的新能峰,其結(jié)合能為400.2eV,其對(duì)應(yīng)的是質(zhì)子化的氮原子(N+),說(shuō)明改性殼聚糖過(guò)程中加入的酸使得部分氨基質(zhì)子化了,但吸附劑吸附Cl后其質(zhì)子化的氮原子減少,可能是質(zhì)子化的殼聚糖帶正電,其表面的部分電荷與溶液中帶負(fù)電的SO42-和Cl發(fā)生了電中和。CTS的C1s圖譜峰值對(duì)應(yīng)的結(jié)合能284.6、286.2、288.34eV分別屬于基團(tuán)C—C/C—H、C—O/C—N+、C=O/O—C—O。CTS的N和C—N的結(jié)合能在Hg(Ⅱ)改性后,由399.4、286.2eV移動(dòng)到399.83、296.53eV,說(shuō)明—NH2參與了Hg(Ⅱ)的吸附過(guò)程。Hg-CTS吸附Cl前后S2p光譜的結(jié)合能基本沒(méi)有變化,強(qiáng)度變小。CTS-Hg與CTS能譜圖對(duì)比,出現(xiàn)Hg4f峰,說(shuō)明CTS在改性后成功絡(luò)合上了Hg(Ⅱ)。CTS-Hg吸附Cl前后的能譜圖對(duì)比發(fā)現(xiàn)Cl2p峰,說(shuō)明CTS-Hg吸附了Cl-,主要是由于Hg(Ⅱ)與Cl-絡(luò)合作用形成了HgCl42-,由于發(fā)生了化學(xué)反應(yīng),在吸附劑上形成了較穩(wěn)定的氯汞化合物。
2.1.4 SEM分析
CTS、Hg-CTS和Hg-CTS吸附Cl-后10000倍的SEM見(jiàn)圖2。
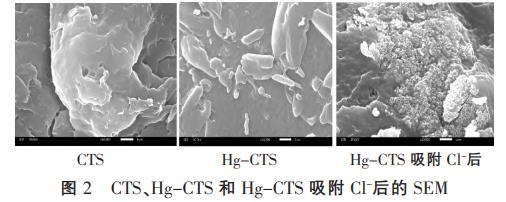
由圖2可知,CTS為非孔材料,呈現(xiàn)出平坦的層狀結(jié)構(gòu),表面光潔無(wú)空洞出現(xiàn);Hg-CTS表面出現(xiàn)塊狀結(jié)構(gòu),且表面緊實(shí)、凹凸不平;Hg-CTS表面在吸附Cl-后變得非常粗糙,結(jié)合XPS表征分析,推測(cè)粗糙面可能為吸附劑表面的氯聚集體。
2.2 Hg-CTS投加量對(duì)Hg-CTS吸附Cl的影響
在20℃,Hg-CTS投加量為0.1~1.0g條件下,分別與100mL1000mg/L的Cl于搖床振蕩30min。考察不同Hg-CTS投加量對(duì)Cl吸附的影響,結(jié)果見(jiàn)圖3。
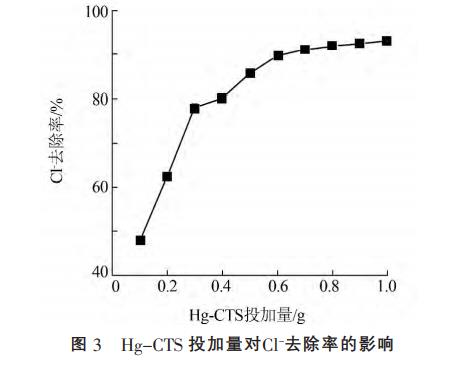
由圖3可知,隨著Hg-CTS投加量增加,Cl去除率逐漸增大,0.8g后繼續(xù)增大投加量,Cl去除率變化不大。這是由于在Cl初始濃度一定情況下,隨著HgCST投加量的增加,Hg-CST上活性位點(diǎn)的可用性隨著投加量增大而增大,去除率也隨著增加。投加量達(dá)0.8g后Cl去除率趨于平衡,這是由于過(guò)多的吸附劑使吸附劑顆粒之間相互聚合,阻礙了吸附劑對(duì)Cl的吸附作用。當(dāng)吸附劑投加量為8.0g/L時(shí),Cl-去除率為91.91%,出水Cl質(zhì)量濃度為80.9mg/L,遠(yuǎn)低于《工業(yè)用水水質(zhì)》(GB/T19923—2005)回用標(biāo)準(zhǔn)限值250mg/L,綜合考慮成本和去除效果,最佳投加量取8.0g/L。
2.3 溶液初始pH對(duì)Hg-CTS吸附Cl的影響
在20℃,調(diào)節(jié)10組100mL1000mg/L的Cl溶液在不同初始pH條件下,含氯溶液與0.8g的HgCTS混合振蕩60min。結(jié)果表明隨著溶液pH升高,Hg-CTS對(duì)Cl的去除率先升高后下降。溶液的初始pH在1~10,吸附劑對(duì)Cl的去除率均大于85.07%,Cl出水濃度均能達(dá)到回用標(biāo)準(zhǔn)。當(dāng)pH為3時(shí),Cl去除率達(dá)到最大95.12%;當(dāng)pH>3時(shí),Cl的去除率略有所下降。由此可以看出吸附劑Hg-CTS具有較寬的pH適應(yīng)范圍。
2.4反應(yīng)時(shí)間對(duì)Hg-CTS吸附Cl的影響
在20℃下,8.0g/LHg-CTS與不同初始濃度的Cl-溶液混合,振蕩一定時(shí)間。考察反應(yīng)時(shí)間對(duì)HgCTS吸附Cl的影響,結(jié)果見(jiàn)圖4。
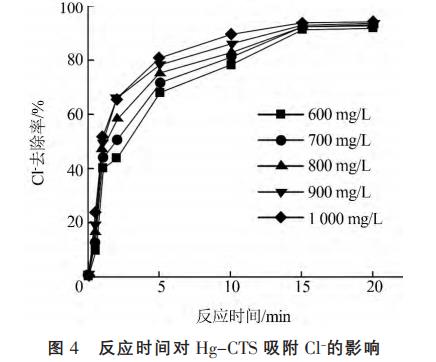
由圖4可知,Hg-CTS對(duì)Cl吸附速率很快,與初始質(zhì)量濃度1000mg/L的Cl反應(yīng)在20min內(nèi)達(dá)到吸附平衡,Cl去除率達(dá)到94%,這是由于Hg-CTS對(duì)Cl有較強(qiáng)的富集能力,使得在短時(shí)間內(nèi)達(dá)到吸附平衡。在前15min內(nèi),吸附速率非常快,此后,隨著吸附時(shí)間的延長(zhǎng),Hg-CTS對(duì)Cl的去除率變化不明顯,且未出現(xiàn)解吸附的現(xiàn)象,因此反應(yīng)時(shí)間取20min為最佳。
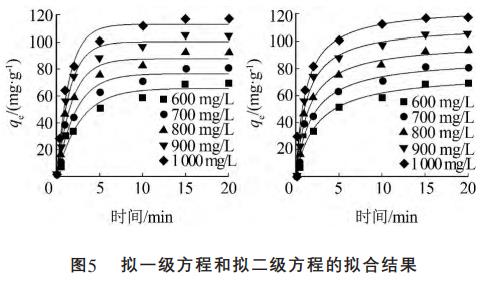
2.5 吸附動(dòng)力學(xué)分析
在吸附時(shí)間0~25min范圍內(nèi),用擬一級(jí)動(dòng)力學(xué)方程和擬二級(jí)動(dòng)力學(xué)方程對(duì)圖2中的數(shù)據(jù)進(jìn)行擬合。表達(dá)式見(jiàn)式(1)、式(2)。
式中:
qt———t時(shí)刻的吸附量,mg/g;
qe——吸附平衡時(shí)的吸附量,mg/g;
k1——擬一級(jí)動(dòng)力學(xué)方程吸附速率常數(shù),min-1;
k2——擬二級(jí)動(dòng)力學(xué)方程吸附速率常數(shù),g/(mg·min)。
動(dòng)力學(xué)方程非線(xiàn)性擬合結(jié)果見(jiàn)圖5。
式(1)和式(2)對(duì)圖2中的數(shù)據(jù)進(jìn)行擬合得到的動(dòng)力學(xué)相關(guān)參數(shù)見(jiàn)表1。
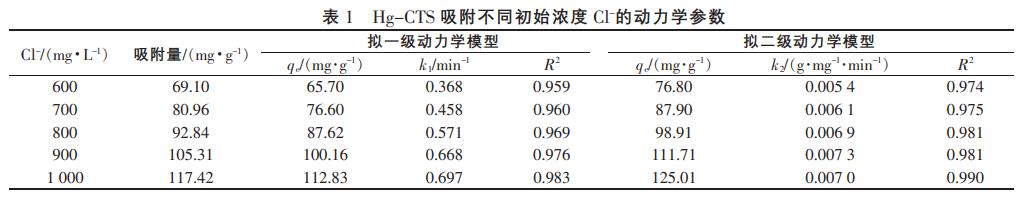
由圖5、表1可知,擬合曲線(xiàn)和R2表明,兩個(gè)模型都能較好地描述Hg-CTS對(duì)Cl的吸附過(guò)程。對(duì)比擬合結(jié)果還發(fā)現(xiàn),擬二級(jí)動(dòng)力學(xué)模型比擬一級(jí)動(dòng)力學(xué)模型更適合用于闡釋Hg-CTS對(duì)Cl的吸附過(guò)程。擬二級(jí)動(dòng)力學(xué)模型是以化學(xué)吸附為基礎(chǔ)的,因此吸附過(guò)程中可能發(fā)生了化學(xué)反應(yīng)。根據(jù)擬二級(jí)動(dòng)力學(xué)方程可以求出Cl-在Hg-CTS吸附劑上的吸附速率常數(shù),當(dāng)Cl初始質(zhì)量濃度為1000mg/L時(shí),吸附量達(dá)到117.42mg/g。其吸附量大于常用樹(shù)脂、水滑石的吸附量。
2.6 共存離子對(duì)處理實(shí)際廢水的影響
為了嘗試所制備的吸附劑對(duì)實(shí)際硫酸廢水的處理效果及共存離子SO42-對(duì)吸附的影響,選取了廣西某冶煉廠的硫酸廢水和投加Na2SO4增大廢水SO42-濃度進(jìn)行了除氯實(shí)驗(yàn)。在20℃下,8.0g/LHg-CTS與兩份Cl-質(zhì)量濃度為1050mg/L的硫酸廢水反應(yīng),其中廢水的ρ(SO42-)=1.84g/L,競(jìng)爭(zhēng)實(shí)驗(yàn)廢水投加Na2SO4后溶液中ρ(SO42-)=3.38g/L。一段時(shí)間后取上清液測(cè)定Cl-含量,60min前Hg-CTS對(duì)硫酸廢水中Cl吸附速率較快,60min時(shí)Cl-去除率達(dá)到79.65%,隨著反應(yīng)時(shí)間的增加,吸附速率逐漸變緩慢,在120min時(shí)達(dá)到吸附平衡,Cl去除率達(dá)到90.39%,廢水中殘留Cl-為100.91mg/L,低于《工業(yè)用水水質(zhì)》(GB/T19923—2005)回用標(biāo)準(zhǔn)限值250mg/L。同時(shí)競(jìng)爭(zhēng)實(shí)驗(yàn)結(jié)果表明,SO42-對(duì)Hg-CTS吸附氯離子基本沒(méi)影響,Hg-CTS的吸附具有很好的離子選擇性。
3、結(jié)論
(1)通過(guò)將硫酸汞與兩種有機(jī)酸形成的配位化合物與殼聚糖氨基反應(yīng)后,制備出高效Hg-CTS吸附劑,在Cl-初始質(zhì)量濃度為1000mg/L,投加量8.0g/L,時(shí)間20min,pH為3條件下,Cl-的去除率達(dá)到94%,吸附量為117.42mg/g。同時(shí),Hg-CTS吸附劑對(duì)Cl-吸附動(dòng)力學(xué)符合擬二級(jí)動(dòng)力學(xué)模型。
(2)該吸附劑對(duì)Cl-表現(xiàn)出良好的吸附性能,對(duì)廣西某冶煉廠硫酸廢水中Cl-去除率達(dá)到90%以上,Cl-殘留濃度低于《工業(yè)用水水質(zhì)》(GB/T19923—2005)回用標(biāo)準(zhǔn)限值250mg/L。
(3)從吸附劑表征結(jié)果發(fā)現(xiàn),Cl-的去除主要是靠吸附劑上Hg2+對(duì)Cl-的富集來(lái)實(shí)現(xiàn)。
廣東建樹(shù)環(huán)保科技有限公司是一家專(zhuān)業(yè)從事工業(yè)廢水處理、工業(yè)廢氣處理和環(huán)境修復(fù)的環(huán)保設(shè)備研發(fā)與銷(xiāo)售服務(wù)的企業(yè)。為工業(yè)企業(yè)和市政工程等項(xiàng)目提供工業(yè)廢水處理、工業(yè)廢氣處理、有機(jī)廢氣VOCs處理的一體化解決方案,從“工程設(shè)計(jì)”、“工程承包”、“設(shè)備采購(gòu)”、“安裝調(diào)試”、“耗材銷(xiāo)售”、“運(yùn)營(yíng)管理”、“環(huán)評(píng)辦理”等環(huán)節(jié)提供專(zhuān)業(yè)的差異化服務(wù),聯(lián)系電話(huà):135 5665 1700。





